03 - Create a basic workflow
Table of contents
- 03 - Create a basic workflow
- Table of contents
- Takeaways
- Summary commands
- Our final snakemake workflow!
3.1 Aim
Let’s create a basic workflow that will do some of the typical quality control checks, pre-processing and mapping to a reference genome that is undertaken on paired-end sequence data
We have paired end sequencing data for three samples NA24631 to process in the ./data directory. Let’s have a look:
ls -lh ./data/
Output:
-rw-rw-r-- 1 lkemp lkemp 2.1M Nov 18 14:56 NA24631_1.fastq.gz
-rw-rw-r-- 1 lkemp lkemp 2.3M Nov 18 14:56 NA24631_2.fastq.gz
-rw-rw-r-- 1 lkemp lkemp 2.1M Nov 18 14:56 NA24694_1.fastq.gz
-rw-rw-r-- 1 lkemp lkemp 2.3M Nov 18 14:56 NA24694_2.fastq.gz
-rw-rw-r-- 1 lkemp lkemp 1.8M Nov 18 14:56 NA24695_1.fastq.gz
-rw-rw-r-- 1 lkemp lkemp 1.9M Nov 18 14:56 NA24695_2.fastq.gz
3.2 File structure
Workflow file structure:
demo_workflow/
|_______results/
|_______workflow/
|_______envs/
|_______Snakefile
We will work in the workflow directory send all of our file outputs/results to the results/ directory
Read up on the best practice workflow structure here
Create this file structure and our main Snakefile with:
mkdir -p demo_workflow/{results,workflow/envs}
touch demo_workflow/workflow/Snakefile
3.3 First rule
First lets run the first step (fastqc) directly on the command line to get the syntax of the command right and check what outputs files we expect to get
# Install fastqc
conda install fastqc
# See what parameters are available
fastqc --help
# Create a test directory
mkdir test
# Directly run fastqc on the command line
fastqc ./data/NA24631_1.fastq.gz ./data/NA24631_2.fastq.gz -o ./test -t 8
What are the output files of fastqc? Find out with:
ls -lh ./test
My output:
-rw-rw-r-- 1 lkemp lkemp 250K Nov 18 15:53 NA24631_1_fastqc.html
-rw-rw-r-- 1 lkemp lkemp 327K Nov 18 15:53 NA24631_1_fastqc.zip
-rw-rw-r-- 1 lkemp lkemp 249K Nov 18 15:53 NA24631_2_fastqc.html
-rw-rw-r-- 1 lkemp lkemp 327K Nov 18 15:53 NA24631_2_fastqc.zip
Let’s wrap this up in a Snakemake workflow! Start with the basic structure in the Snakefile:
# Target OUTPUT files for the whole workflow
rule all:
input:
# Workflow
rule my_rule:
input:
""
output:
""
threads:
shell:
""
- Name the rule
- Fill in the the input fastq files from the
datadirectory (path relative to the Snakefile) - Fill in the output files (now you can see it’s useful to know what files fasqtc outputs!)
- Set the number of threads
- Write the shell command and pass these variables to the shell command
- Set the final output files (
rule all:)
# Target OUTPUT files for the whole workflow
rule all:
input:
+ "../results/fastqc/NA24631_1_fastqc.html",
+ "../results/fastqc/NA24631_2_fastqc.html",
+ "../results/fastqc/NA24631_1_fastqc.zip",
+ "../results/fastqc/NA24631_2_fastqc.zip"
# Workflow
+ rule fastqc:
input:
+ R1 = "../../data/NA24631_1.fastq.gz",
+ R2 = "../../data/NA24631_2.fastq.gz"
output:
+ html = ["../results/fastqc/NA24631_1_fastqc.html", "../results/fastqc/NA24631_2_fastqc.html"],
+ zip = ["../results/fastqc/NA24631_1_fastqc.zip", "../results/fastqc/NA24631_2_fastqc.zip"]
+ threads: 8
shell:
+ "fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads}"
Let’s test the workflow! First we need to be in the workflow directory, where the Snakefile is
cd demo_workflow/workflow/
Then let’s carry out a dryrun of the workflow, where no actual analysis is undertaken (fastqc is not run) but the overall Snakemake structure is run/validated. This is a good way to check for errors in your Snakemake workflow before actually running your workflow.
snakemake --dryrun --cores 8
Output:
Building DAG of jobs...
Job counts:
count jobs
1 all
1 fastqc
2
[Wed Nov 18 17:08:08 2020]
rule fastqc:
input: ../../data/NA24631_1.fastq.gz, ../../data/NA24631_2.fastq.gz
output: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 1
threads: 8
[Wed Nov 18 17:08:08 2020]
localrule all:
input: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 0
Job counts:
count jobs
1 all
1 fastqc
2
This was a dry-run (flag --dryrun). The order of jobs does not reflect the order of execution.
The output confirms that the workflow will run one sample (count 1) through jobs fastqc
We can also visualise our workflow by creating a directed acyclic graph (DAG). We tell snakemake to create a DAG with the --dag flag, then pipe this output to the dot software and write the output to the file dag_1.png
snakemake --dag | dot -Tpng > dag_1.png
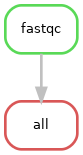
Our diagram has a node for each job which are connected by edges representing dependencies
Note. this diagram can be output to several other image formats such as svg or pdf
Let’s do a full run of our workflow (by removing the --dryrun flag)
snakemake --cores 8
Output:
Building DAG of jobs...
Using shell: /bin/bash
Provided cores: 8
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1 fastqc
2
[Fri Nov 20 18:46:27 2020]
rule fastqc:
input: ../../data/NA24631_1.fastq.gz, ../../data/NA24631_2.fastq.gz
output: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 1
threads: 8
Started analysis of NA24631_1.fastq.gz
Approx 5% complete for NA24631_1.fastq.gz
Approx 10% complete for NA24631_1.fastq.gz
Approx 15% complete for NA24631_1.fastq.gz
Approx 20% complete for NA24631_1.fastq.gz
Approx 25% complete for NA24631_1.fastq.gz
Approx 30% complete for NA24631_1.fastq.gz
Approx 35% complete for NA24631_1.fastq.gz
Approx 40% complete for NA24631_1.fastq.gz
Approx 45% complete for NA24631_1.fastq.gz
Approx 50% complete for NA24631_1.fastq.gz
Approx 55% complete for NA24631_1.fastq.gz
Started analysis of NA24631_2.fastq.gz
Approx 60% complete for NA24631_1.fastq.gz
Approx 65% complete for NA24631_1.fastq.gz
Approx 5% complete for NA24631_2.fastq.gz
Approx 10% complete for NA24631_2.fastq.gz
Approx 70% complete for NA24631_1.fastq.gz
Approx 75% complete for NA24631_1.fastq.gz
Approx 15% complete for NA24631_2.fastq.gz
Approx 20% complete for NA24631_2.fastq.gz
Approx 80% complete for NA24631_1.fastq.gz
Approx 25% complete for NA24631_2.fastq.gz
Approx 85% complete for NA24631_1.fastq.gz
Approx 30% complete for NA24631_2.fastq.gz
Approx 90% complete for NA24631_1.fastq.gz
Approx 35% complete for NA24631_2.fastq.gz
Approx 95% complete for NA24631_1.fastq.gz
Approx 40% complete for NA24631_2.fastq.gz
Analysis complete for NA24631_1.fastq.gz
Approx 45% complete for NA24631_2.fastq.gz
Approx 50% complete for NA24631_2.fastq.gz
Approx 55% complete for NA24631_2.fastq.gz
Approx 60% complete for NA24631_2.fastq.gz
Approx 65% complete for NA24631_2.fastq.gz
Approx 70% complete for NA24631_2.fastq.gz
Approx 75% complete for NA24631_2.fastq.gz
Approx 80% complete for NA24631_2.fastq.gz
Approx 85% complete for NA24631_2.fastq.gz
Approx 90% complete for NA24631_2.fastq.gz
Approx 95% complete for NA24631_2.fastq.gz
Analysis complete for NA24631_2.fastq.gz
[Fri Nov 20 18:46:33 2020]
Finished job 1.
1 of 2 steps (50%) done
[Fri Nov 20 18:46:33 2020]
localrule all:
input: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 0
[Fri Nov 20 18:46:33 2020]
Finished job 0.
2 of 2 steps (100%) done
Complete log: /home/lkemp/RezBaz2020_snakemake_workshop/demo_workflow/workflow/.snakemake/log/2020-11-20T184627.461379.snakemake.log
It worked! Now in our results directory we have our output files from fastqc. Let’s have a look:
ls -lh ../results/fastqc/
Output
total 2.4M
-rw-rw-r-- 1 lkemp lkemp 718K Nov 20 18:46 NA24631_1_fastqc.html
-rw-rw-r-- 1 lkemp lkemp 475K Nov 20 18:46 NA24631_1_fastqc.zip
-rw-rw-r-- 1 lkemp lkemp 726K Nov 20 18:46 NA24631_2_fastqc.html
-rw-rw-r-- 1 lkemp lkemp 479K Nov 20 18:46 NA24631_2_fastqc.zip
What happens if we try a dryrun or full run now?
snakemake --dryrun --cores 8
snakemake --cores 8
Output
Building DAG of jobs...
Nothing to be done.
Nothing happens, all the target files in rule all have already been created so Snakemake does nothing
Also, what happens if we create another directed acyclic graph (DAG) after the workflow has been run?
snakemake --dag | dot -Tpng > dag.png
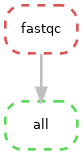
Notice our workflow ‘job nodes’ are now dashed lines, this indicates that their output is up to date and therefore the rule doesn’t need to be run. We already have our target files!
This can be quite informative if your workflow errors out at a rule. You can visually check which rules successfully ran and which didn’t.
3.4 Run using the conda package management system
fastqc worked because we already had it installed locally. Let’s specify a conda environment for fastqc so the user of the workflow doesn’t need to install it manually.
Make a conda environment file for fastqc
# Create the file
touch ./envs/fastqc.yaml
# See what versions of fastqc are available
conda search fastqc
# Write the following to fastqc.yaml
channels:
- bioconda
- conda-forge
- defaults
dependencies:
- bioconda::fastqc=0.11.9
This will install fastqc (version 0.11.9) from bioconda into a ‘clean’ conda environment separate from the rest of your computer
Have a look at bioconda’s list of packages to see the VERY extensive list of open source (free) bioinformatics software that is available for download and use. Note that is only one of the conda package repositories that exist, also have a look at the conda-forge and main conda package repositories.
See here for information on creating conda environment files.
Update our rule to use it using the conda: directive
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/fastqc/NA24631_1_fastqc.html",
"../results/fastqc/NA24631_2_fastqc.html",
"../results/fastqc/NA24631_1_fastqc.zip",
"../results/fastqc/NA24631_2_fastqc.zip"
# Workflow
rule fastqc:
input:
R1 = "../../data/NA24631_1.fastq.gz",
R2 = "../../data/NA24631_2.fastq.gz"
output:
html = ["../results/fastqc/NA24631_1_fastqc.html", "../results/fastqc/NA24631_2_fastqc.html"],
zip = ["../results/fastqc/NA24631_1_fastqc.zip", "../results/fastqc/NA24631_2_fastqc.zip"]
threads: 8
+ conda:
+ "envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads}"
Run again, now telling Snakemake to use to use Conda to get our software by using the --use-conda flag
# Remove output of last run
rm -r ../results/*
# Run dryrun again
- snakemake --dryrun --cores 8
+ snakemake --dryrun --cores 8 --use-conda
My output:
Building DAG of jobs...
Conda environment envs/fastqc.yaml will be created.
Job counts:
count jobs
1 all
1 fastqc
2
[Thu Nov 26 16:10:03 2020]
rule fastqc:
input: ../../data/NA24631_1.fastq.gz, ../../data/NA24631_2.fastq.gz
output: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 1
threads: 8
[Thu Nov 26 16:10:03 2020]
localrule all:
input: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
jobid: 0
Job counts:
count jobs
1 all
1 fastqc
2
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
Notice it now says that “Conda environment envs/fastqc.yaml will be created.”. Now the software our workflow uses will be automatically installed!
# Run again
- snakemake --cores 8
+ snakemake --cores 8 --use-conda
3.5 Capture our logs
So far our logs (for fastqc) have been simply printed to our screen. As you can imagine, if you had a large automated workflow (that you might not be sitting at the computer watching run) you’ll want to capture all that information. Therefore, any information the software spits out (including error messages!) will be kept and can be looked at once you return to your machine from your coffee break.
We can get the logs for each rule to be written to a log file via the log: directive:
- It’s a good idea to organise the logs by:
- Putting the logs in a directory labelled after the rule/software that was run
- Labelling the log files with the sample name the software was run on
- Also make sure you tell the software (fastqc) to write the standard output and standard error to this log file we defined in the
log:directive in the shell script (eg.&> {log})
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/fastqc/NA24631_1_fastqc.html",
"../results/fastqc/NA24631_2_fastqc.html",
"../results/fastqc/NA24631_1_fastqc.zip",
"../results/fastqc/NA24631_2_fastqc.zip"
# Workflow
rule fastqc:
input:
R1 = "../../data/NA24631_1.fastq.gz",
R2 = "../../data/NA24631_2.fastq.gz"
output:
html = ["../results/fastqc/NA24631_1_fastqc.html", "../results/fastqc/NA24631_2_fastqc.html"],
zip = ["../results/fastqc/NA24631_1_fastqc.zip", "../results/fastqc/NA24631_2_fastqc.zip"]
+ log:
+ "logs/fastqc/NA24631.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
- "fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads}"
+ "fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
A tangent about standard streams
- These are standard streams in which information is returned by a computer process - in our case the logs that we see returned to us on our screen when we run fastqc
- There are two main streams:
- standard output (the log messages)
- standard error (the error messages)
Different ways to write log files:
| Syntax | standard output in terminal | standard error in terminal | standard output in file | standard error in file |
|---|---|---|---|---|
> |
:x: | :heavy_check_mark: | :heavy_check_mark: | :x: |
2> |
:heavy_check_mark: | :x: | :x: | :heavy_check_mark: |
&> |
:x: | :x: | :heavy_check_mark: | :heavy_check_mark: |
(Table adapted from here)
Exercise:
Try creating an error in the shell command (for example remove the
-oflag) and use the three different syntaxes for writing to your log file. What is and isn’t printed to your screen and to your log file?
Like this? Read some more
Run again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
We now have a log file, lets have a look at the first 10 lines of our log with:
head ./logs/fastqc/NA24631.log
Output:
Started analysis of NA24631_1.fastq.gz
Approx 5% complete for NA24631_1.fastq.gz
Approx 10% complete for NA24631_1.fastq.gz
Approx 15% complete for NA24631_1.fastq.gz
Approx 20% complete for NA24631_1.fastq.gz
Approx 25% complete for NA24631_1.fastq.gz
Approx 30% complete for NA24631_1.fastq.gz
Approx 35% complete for NA24631_1.fastq.gz
Approx 40% complete for NA24631_1.fastq.gz
Approx 45% complete for NA24631_1.fastq.gz
We have logs. Tidy logs.

3.6 Scale up to analyse all of our samples
We are currently only analysing one of our three samples
Let’s scale up to run all of our samples by using wildcards, this way we can grab all the samples/files in the data directory and analyse them
- Set a global wildcard that defines the samples to be analysed
- Generalise where this rule uses an individual sample (
NA24631) to use this wildcard{sample} - Use the expand function (
expand()) function to tell snakemake that{sample}is what we defined in our global wildcardSAMPLES, - Snakemake can figure out what
{sample}is in our rule since it’s defined in the targets inrule all:
# Define samples from data directory using wildcards
+ SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
- "../results/fastqc/NA24631_1_fastqc.html",
- "../results/fastqc/NA24631_2_fastqc.html",
- "../results/fastqc/NA24631_1_fastqc.zip",
- "../results/fastqc/NA24631_2_fastqc.zip"
+ expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES)
# Workflow
rule fastqc:
input:
- R1 = "../../data/NA24631_1.fastq.gz",
- R2 = "../../data/NA24631_2.fastq.gz"
+ R1 = "../../data/{sample}_1.fastq.gz",
+ R2 = "../../data/{sample}_2.fastq.gz"
output:
- html = ["../results/fastqc/NA24631_1_fastqc.html", "../results/fastqc/NA24631_2_fastqc.html"],
- zip = ["../results/fastqc/NA24631_1_fastqc.zip", "../results/fastqc/NA24631_2_fastqc.zip"]
+ html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
+ zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
- "logs/fastqc/NA24631.log"
+ "logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
Visualise workflow
snakemake --dag | dot -Tpng > dag_2.png
Now we have three samples running though our workflow
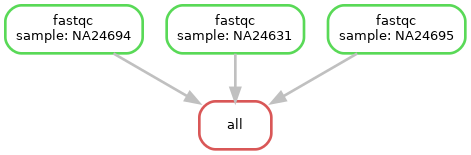
Run workflow again
# Remove output of last run
rm -r ../results/*
# Run dryrun again
snakemake --dryrun --cores 8 --use-conda
See how it now runs each sample over all three of our samples in the output of the dryrun:
Building DAG of jobs...
Job counts:
count jobs
1 all
3 fastqc
4
[Thu Nov 26 22:43:18 2020]
rule fastqc:
input: ../../data/NA24631_1.fastq.gz, ../../data/NA24631_2.fastq.gz
output: ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip
log: logs/fastqc/NA24631.log
jobid: 2
wildcards: sample=NA24631
threads: 8
[Thu Nov 26 22:43:18 2020]
rule fastqc:
input: ../../data/NA24695_1.fastq.gz, ../../data/NA24695_2.fastq.gz
output: ../results/fastqc/NA24695_1_fastqc.html, ../results/fastqc/NA24695_2_fastqc.html, ../results/fastqc/NA24695_1_fastqc.zip, ../results/fastqc/NA24695_2_fastqc.zip
log: logs/fastqc/NA24695.log
jobid: 3
wildcards: sample=NA24695
threads: 8
[Thu Nov 26 22:43:18 2020]
rule fastqc:
input: ../../data/NA24694_1.fastq.gz, ../../data/NA24694_2.fastq.gz
output: ../results/fastqc/NA24694_1_fastqc.html, ../results/fastqc/NA24694_2_fastqc.html, ../results/fastqc/NA24694_1_fastqc.zip, ../results/fastqc/NA24694_2_fastqc.zip
log: logs/fastqc/NA24694.log
jobid: 1
wildcards: sample=NA24694
threads: 8
[Thu Nov 26 22:43:18 2020]
localrule all:
input: ../results/fastqc/NA24694_1_fastqc.html, ../results/fastqc/NA24631_1_fastqc.html, ../results/fastqc/NA24695_1_fastqc.html, ../results/fastqc/NA24694_2_fastqc.html, ../results/fastqc/NA24631_2_fastqc.html, ../results/fastqc/NA24695_2_fastqc.html, ../results/fastqc/NA24694_1_fastqc.zip, ../results/fastqc/NA24631_1_fastqc.zip, ../results/fastqc/NA24695_1_fastqc.zip, ../results/fastqc/NA24694_2_fastqc.zip, ../results/fastqc/NA24631_2_fastqc.zip, ../results/fastqc/NA24695_2_fastqc.zip
jobid: 0
Job counts:
count jobs
1 all
3 fastqc
4
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
# Run again
snakemake --cores 8 --use-conda
All three samples were run through our workflow! And we have a log file for each sample for the fastqc rule
ls -lh ./logs/fastqc
Output:
-rw-rw-r-- 1 lkemp lkemp 1.8K Nov 19 15:17 NA24631.log
-rw-rw-r-- 1 lkemp lkemp 1.8K Nov 19 15:17 NA24694.log
-rw-rw-r-- 1 lkemp lkemp 1.8K Nov 19 15:17 NA24695.log
3.7 Add more rules
- Make a conda environment file for multiqc
# Create the file
touch ./envs/multiqc.yaml
# See what versions of multiqc are available
conda search multiqc
# Write the following to multiqc.yaml
channels:
- bioconda
- conda-forge
- defaults
dependencies:
- bioconda::multiqc=1.9
- Connect the outputs of fastqc to the inputs of multiqc
- Add a new final target for
rule all:
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES),
+ "../results/multiqc_report.html"
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
+ rule multiqc:
+ input:
+ ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
+ output:
+ "../results/multiqc_report.html"
+ log:
+ "logs/multiqc/multiqc.log"
+ conda:
+ "envs/multiqc.yaml"
+ shell:
+ "multiqc {input} -o ../results/ &> {log}"
Run workflow again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
Didn’t work? Error:
Building DAG of jobs...
WildcardError in line 30 of /home/lkemp/RezBaz2020/demo_workflow/workflow/Snakefile:
Wildcards in input files cannot be determined from output files:
'sample'
Since we haven’t defined {sample} in rule all: for multiqc, we need to define it somewhere! Let do so in the multiqc rule
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES),
"../results/multiqc_report.html"
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
- ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
+ expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
Visualise workflow
snakemake --dag | dot -Tpng > dag_3.png
Now we have two rules in our workflow (fastqc and multiqc), we can also see that multiqc isn’t run for each sample (since it merges the output of fastqc for all samples)
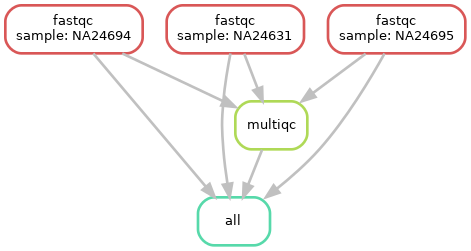
Run again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
What happens if we only have the final target file (../results/multiqc_report.html) in rule all:
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
- expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
- expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
- expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
- expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES),
"../results/multiqc_report.html"
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
Run workflow again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
It still works because it is the last file in the workflow sequence, Snakemake will do all the steps necessary to get to this target file (therefore it runs fastqc and multiqc)
Visualise workflow
snakemake --dag | dot -Tpng > dag_4.png
Although the workflow ran the same, the DAG actually changed slightly, now there is only one file target and only the output of multiqc goes to rule all
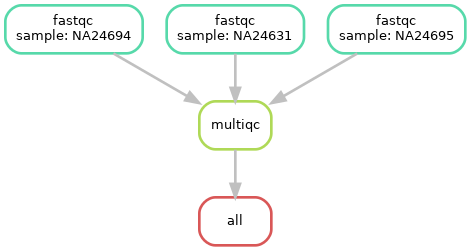
Beware: Snakemake will also NOT run rules that is doesn’t need to run in order to get the target files defined in rule: all
For example if only our fastqc outputs are defined as the target in rule: all
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
+ expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
+ expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES)
- "../results/multiqc_report.html"
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
Run again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
Output:
Job counts:
count jobs
1 all
3 fastqc
4
This was a dry-run (flag --dryrun). The order of jobs does not reflect the order of execution.
Our multiqc rule won’t be run/evaluated
Visualise workflow
snakemake --dag | dot -Tpng > dag_5.png
Now we are back to only running fastqc in our workflow, despite having our second rule (multiqc) in our workflow
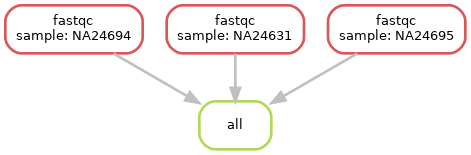
Snakemake is lazy.

3.8 Add even more rules
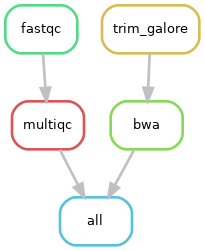
Let’s add the rest of the rules. We want to get to:
We currently have fastqc and multiqc, so we still need to add trim_galore and bwa
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
- expand("../results/fastqc/{sample}_1_fastqc.html", sample = SAMPLES),
- expand("../results/fastqc/{sample}_2_fastqc.html", sample = SAMPLES),
- expand("../results/fastqc/{sample}_1_fastqc.zip", sample = SAMPLES),
- expand("../results/fastqc/{sample}_2_fastqc.zip", sample = SAMPLES)
+ "../results/multiqc_report.html",
+ expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
+ rule trim_galore:
+ input:
+ ["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
+ output:
+ ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
+ log:
+ "logs/trim_galore/{sample}.log"
+ conda:
+ "./envs/trim_galore.yaml"
+ threads: 8
+ shell:
+ "trim_galore {input} -o ../results/trimmed/ --paired --cores {threads} &> {log}"
+ rule bwa:
+ input:
+ fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
+ refgenome = "/store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta"
+ output:
+ "../results/mapped/{sample}.bam"
+ log:
+ "logs/bwa_mem/{sample}.log"
+ conda:
+ "./envs/bwa.yaml"
+ threads: 8
+ shell:
+ "bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
Create conda env files
# Create file
touch ./envs/trim_galore.yaml
# Write the following to trim_galore.yaml
channels:
- bioconda
- conda-forge
- defaults
dependencies:
- bioconda::trim-galore=0.6.5
# Create file
touch ./envs/bwa.yaml
# Write the following to bwa.yaml
channels:
- bioconda
- conda-forge
- defaults
dependencies:
- bioconda::bwa=0.7.17
- bioconda::gatk4=4.1.6.0
Visualise workflow
snakemake --dag | dot -Tpng > dag_6.png
Fantastic, we are starting to build a workflow!
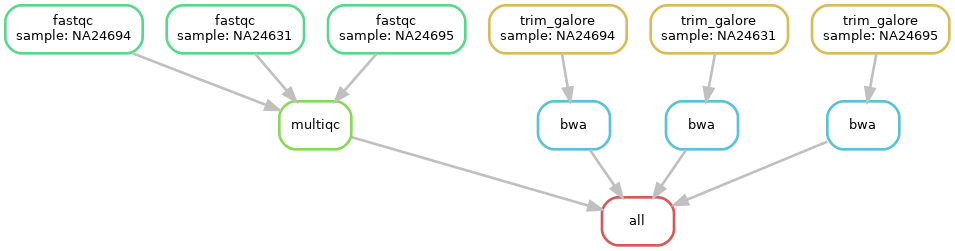
However, when analysing many samples, our DAG can become messy and complicated. Instead, we can create a rulegraph that will let us visualise our workflow without showing every single sample that will run through it
snakemake --rulegraph | dot -Tpng > rulegraph_1.png
An aside: another option that will show all your input and output files at each step:
snakemake --filegraph | dot -Tpng > filegraph.png
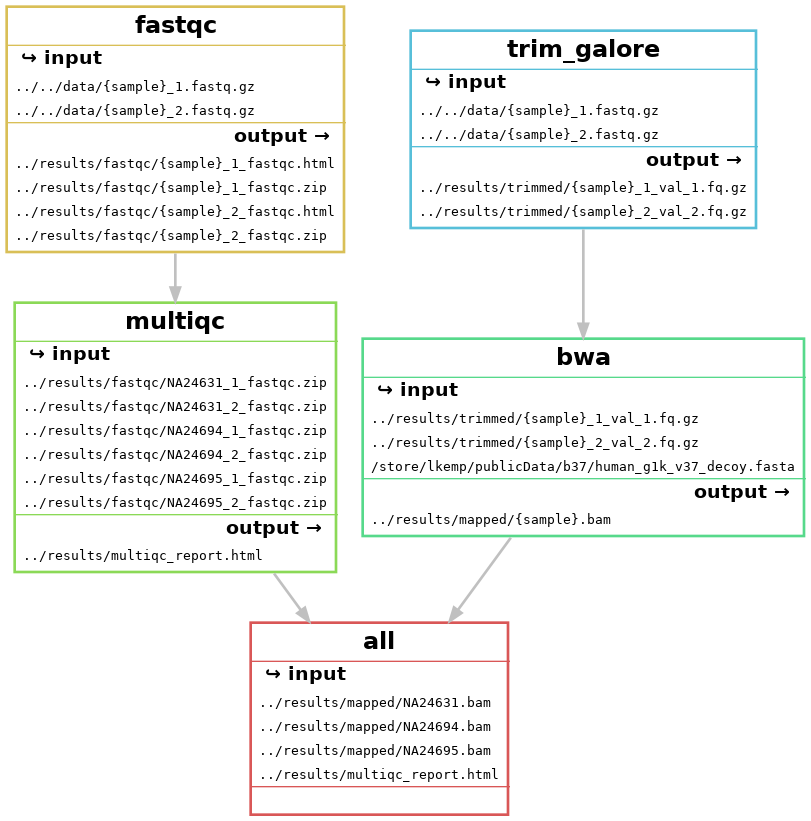
Run the workflow again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 8 --use-conda
snakemake --cores 8 --use-conda
Notice it will run only one rule/sample at a time…why is that?
3.9 Throw it more cores
Run again allowing Snakemake to use more cores overall --cores 32 rather than --cores 8 (only if you are working on a machine with this many cores! Your laptop may well not!)
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 32 --use-conda
snakemake --cores 32 --use-conda
Notice it ran alot faster and there were more samples and rules running at one time. This is because we set each rule to run with 8 threads. Initially we specified that the maximum number of cores to be used by the workflow was 8 with the --cores 8 flag, meaning only one rule and sample can be run at one time. When we increased the maximum number of cores to be used by the workflow to 32 with --cores 32, up to 4 samples could be run through .
With a high performance cluster such as NeSi, you can start to REALLY scale up.
Boom! Scalability here we come!

Takeaways
- There is an enormous range of open source (free) software available for use from the bioconda, conda-forge and main conda package repositories
- These software are all very straightforward to integrate in your snakemake workflow
- Run your commands directly on the command line before wrapping it up in a Snakemake rule
- First do a dryrun to check the Snakemake structure is set up correctly
- Work iteratively (get each rule working before moving onto the next)
- File paths are relative to the Snakefile
- Run your workflow from where your Snakefile is
- Visualise your workflow by creating a DAG (directed acyclic graph), a rulegraph or filegraph
- Use conda to install software in your workflow - this improves reproducibility and portability
- Snakemake is lazy…
- It will only do something if it hasn’t already done it
- It will pick up where it left off, rather than run the whole workflow again
- It won’t do any steps that aren’t necessary to get to the target files defined in
rule: all
input:output:log:andthreads:directives need to be called in theshelldirective- Capture your log files
- Organise your log files by naming them after the rule that was run and sample that was analysed
- You don’t need to specify all the target files in
rule all:, the final file in a given chain of tasks will suffice - We can massively speed up our analyses by running our samples in parallel
Summary commands
Create a directed acyclic graph (DAG) with:
snakemake --dag | dot -Tpng > dag.png
Create a rulegraph with:
snakemake --rulegraph | dot -Tpng > rulegraph.png
Create a filegraph with:
snakemake --filegraph | dot -Tpng > filegraph.png
Run a dryrun of your snakemake workflow with:
snakemake --dryrun --cores 8
Run your snakemake workflow with:
snakemake --cores 8
Run a dryrun of your snakemake workflow (using conda to install your software) with:
snakemake --dryrun --cores 8 --use-conda
Run your snakemake workflow (using conda to install your software) with:
snakemake --cores 8 --use-conda
Create a global wildcard to get process all your samples in a directory with:
SAMPLES, = glob_wildcards("../relative/path/to/samples/{sample}_1.fastq.gz")
Combine this with the expand function to tell Snakemake to look at your global wildcard to figure out what you refer to as {sample} in your workflow
expand("../results/{sample}.bam", sample = SAMPLES)
Increase the number of samples that can be analysed at one time in your workflow by increasing the maximum number of cores with the --cores command
snakemake --cores 32 --use-conda
Our final snakemake workflow!
See basic_demo_workflow for the final Snakemake workflow we’ve created up to this point