04 - Leveling up your workflow!
Table of contents
- 04 - Leveling up your workflow!
- Table of contents
- Takeaways
- Summary commands
- Our final snakemake workflow!
4.1 Pull out parameters
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/multiqc_report.html",
expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
rule trim_galore:
input:
["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
output:
["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
+ params:
+ "--paired"
log:
"logs/trim_galore/{sample}.log"
conda:
"./envs/trim_galore.yaml"
threads: 8
shell:
- "trim_galore {input} -o ../results/trimmed/ --paired --cores {threads} &> {log}"
+ "trim_galore {input} -o ../results/trimmed/ {params} --cores {threads} &> {log}"
rule bwa:
input:
fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
refgenome = "/store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta"
output:
"../results/mapped/{sample}.bam"
log:
"logs/bwa_mem/{sample}.log"
conda:
"./envs/bwa.yaml"
threads: 8
shell:
"bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
4.2 Pull out user configurable options
We can separate the user configurable options for our workflow away from the workflow. This supports reproducibility by minimising the chance the user makes changes to the core workflow.
Create a configuration file in a new directory config/
File structure:
demo_workflow/
|_______results/
|_______workflow/
| |_______envs/
| |_______Snakefile
|_______config
|_______config.yaml
# Create config directory
mkdir ../config
# Create configuration file
touch ../config/config.yaml
Now we need to pull out a parameter for the user to configure. Let’s get the user to specify where the reference genome to use in the pipeline. In our ../config/config.yaml file create a place for our user to specify the file path to the reference genome on their machine:
# File path to the reference genome (.fasta)
REFGENOME: ""
In the Snakefile, tell Snakemake to grab the variable REFGENOME from ../config/config.yaml
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/multiqc_report.html",
expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
rule trim_galore:
input:
["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
output:
["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
params:
"--paired"
log:
"logs/trim_galore/{sample}.log"
conda:
"./envs/trim_galore.yaml"
threads: 8
shell:
"trim_galore {input} -o ../results/trimmed/ {params} --cores {threads} &> {log}"
rule bwa:
input:
fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
- refgenome = "/store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta"
+ refgenome = config['REFGENOME']
output:
"../results/mapped/{sample}.bam"
log:
"logs/bwa_mem/{sample}.log"
conda:
"./envs/bwa.yaml"
threads: 8
shell:
"bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
Now we don’t have any hard coded file paths in our workflow - supporting portability.
let’s use our configuration file!
Specify the file path to the reference genome (.fasta) in our configuration file
# File path to the reference genome (.fasta)
REFGENOME: "/store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta"
Run workflow again
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 32 --use-conda
snakemake --cores 32 --use-conda
Didn’t work? Error:
KeyError in line 56 of /home/lkemp/RezBaz2020_snakemake_workshop/final_leveled_up_demo_workflow/workflow/Snakefile:
'REFGENOME'
File "/home/lkemp/RezBaz2020_snakemake_workshop/final_leveled_up_demo_workflow/workflow/Snakefile", line 56, in <module>
Snakemake can’t find our ‘Key’ - we haven’t told Snakemake where our config file is. We can do this by passing the location of our config file to the --configfile flag
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
- snakemake --dryrun --cores 32 --use-conda
- snakemake --cores 32 --use-conda
+ snakemake --dryrun --cores 32 --use-conda --configfile ../config/config.yaml
+ snakemake --cores 32 --use-conda --configfile ../config/config.yaml
Alternatively, we can define our config file in our Snakefile in a situation where the configuration file is likely to always be named the same and be in the exact same location ../config/config.yaml and you don’t need the flexibility for the user to specify their own configuration files:
# Define our configuration file
+ configfile: "../config/config.yaml"
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/multiqc_report.html",
expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
shell:
"multiqc {input} -o ../results/ &> {log}"
rule trim_galore:
input:
["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
output:
["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
params:
"--paired"
log:
"logs/trim_galore/{sample}.log"
conda:
"./envs/trim_galore.yaml"
threads: 8
shell:
"trim_galore {input} -o ../results/trimmed/ {params} --cores {threads} &> {log}"
rule bwa:
input:
fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
refgenome = config['REFGENOME']
output:
"../results/mapped/{sample}.bam"
log:
"logs/bwa_mem/{sample}.log"
conda:
"./envs/bwa.yaml"
threads: 8
shell:
"bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
Then we don’t need to specify where the configuration file is on the command line
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
+ snakemake --dryrun --cores 32 --use-conda
+ snakemake --cores 32 --use-conda
- snakemake --dryrun --cores 32 --use-conda --configfile ../config/config.yaml
- snakemake --cores 32 --use-conda --configfile ../config/config.yaml
4.3 Leave messages for the user
We can provide the user of our workflow more information on what is happening at each stage/rule of our workflow via the message: directive. We are able to call many variables such as:
- Input and output files
{input}and{output} - Specific input and output files such as
{input.refgenome} - Our
{params},{log}and{threads}directives
# Define our configuration file
configfile: "../config/config.yaml"
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/multiqc_report.html",
expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
+ message:
+ "Undertaking quality control checks {input}"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
+ message:
+ "Compiling a HTML report for quality control checks. Writing to {output}."
shell:
"multiqc {input} -o ../results/ &> {log}"
rule trim_galore:
input:
["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
output:
["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
params:
"--paired"
log:
"logs/trim_galore/{sample}.log"
+ message:
+ "Trimming using these parameter: {params}. Writing logs to {log}. Using {threads} threads."
conda:
"./envs/trim_galore.yaml"
threads: 8
shell:
"trim_galore {input} -o ../results/trimmed/ {params} --cores {threads} &> {log}"
rule bwa:
input:
fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
refgenome = config['REFGENOME']
output:
"../results/mapped/{sample}.bam"
log:
"logs/bwa_mem/{sample}.log"
+ message:
+ "Mapping sequences against {input.refgenome}"
conda:
"./envs/bwa.yaml"
threads: 8
shell:
"bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 32 --use-conda
snakemake --cores 32 --use-conda
Now our messages are printed to the screen as our workflow runs
[Tue Nov 24 16:05:49 2020]
Job 10: Trimming using these parameter: --paired --cores. Writing logs to logs/trim_galore/NA24695.log. Using 8 threads.
[Tue Nov 24 16:05:49 2020]
Job 3: Undertaking quality control checks ../../data/NA24631_1.fastq.gz ../../data/NA24631_2.fastq.gz
[Tue Nov 24 16:05:49 2020]
Job 2: Undertaking quality control checks ../../data/NA24694_1.fastq.gz ../../data/NA24694_2.fastq.gz
[Tue Nov 24 16:05:49 2020]
Job 4: Undertaking quality control checks ../../data/NA24695_1.fastq.gz ../../data/NA24695_2.fastq.gz
[Tue Nov 24 16:05:59 2020]
Job 6: Trimming using these parameter: --paired --cores. Writing logs to logs/trim_galore/NA24694.log. Using 8 threads.
[Tue Nov 24 16:05:59 2020]
Job 8: Trimming using these parameter: --paired --cores. Writing logs to logs/trim_galore/NA24631.log. Using 8 threads.
[Tue Nov 24 16:05:59 2020]
Job 9: Mapping sequences against /store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta
[Tue Nov 24 16:06:00 2020]
Job 1: Compiling a HTML report for quality control checks. Writing to ../results/multiqc_report.html.
[Tue Nov 24 16:06:05 2020]
Job 7: Mapping sequences against /store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta
[Tue Nov 24 16:06:06 2020]
Job 5: Mapping sequences against /store/lkemp/publicData/b37/human_g1k_v37_decoy.fasta
4.4 Create temporary files
In our workflow, we are likely to be creating files that we don’t want at the end of the day, but are used or produced by our workflow (intermediate files). We can mark such files as temporary so Snakemake will remove the file once it doesn’t need to use it anymore.
For example, we might not want to keep the trimmed version of our fastq data. Let’s have a look at the files currently produced by our workflow with:
ls -lh ../results/trimmed/
My output:
total 12M
-rw-rw-r-- 1 lkemp lkemp 2.9K Nov 24 16:06 NA24631_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.0M Nov 24 16:06 NA24631_1_val_1.fq.gz
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:06 NA24631_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.2M Nov 24 16:06 NA24631_2_val_2.fq.gz
-rw-rw-r-- 1 lkemp lkemp 2.7K Nov 24 16:06 NA24694_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.1M Nov 24 16:06 NA24694_1_val_1.fq.gz
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:06 NA24694_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.2M Nov 24 16:06 NA24694_2_val_2.fq.gz
-rw-rw-r-- 1 lkemp lkemp 2.8K Nov 24 16:05 NA24695_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 1.8M Nov 24 16:05 NA24695_1_val_1.fq.gz
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:05 NA24695_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 1.8M Nov 24 16:05 NA24695_2_val_2.fq.gz
Let’s mark all the trimmed fastq files as temporary in our Snakefile
# Define our configuration file
configfile: "../config/config.yaml"
# Define samples from data directory using wildcards
SAMPLES, = glob_wildcards("../../data/{sample}_1.fastq.gz")
# Target OUTPUT files for the whole workflow
rule all:
input:
"../results/multiqc_report.html",
expand("../results/mapped/{sample}.bam", sample = SAMPLES)
# Workflow
rule fastqc:
input:
R1 = "../../data/{sample}_1.fastq.gz",
R2 = "../../data/{sample}_2.fastq.gz"
output:
html = ["../results/fastqc/{sample}_1_fastqc.html", "../results/fastqc/{sample}_2_fastqc.html"],
zip = ["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"]
log:
"logs/fastqc/{sample}.log"
threads: 8
conda:
"envs/fastqc.yaml"
message:
"Undertaking quality control checks {input}"
shell:
"fastqc {input.R1} {input.R2} -o ../results/fastqc/ -t {threads} &> {log}"
rule multiqc:
input:
expand(["../results/fastqc/{sample}_1_fastqc.zip", "../results/fastqc/{sample}_2_fastqc.zip"], sample = SAMPLES)
output:
"../results/multiqc_report.html"
log:
"logs/multiqc/multiqc.log"
conda:
"envs/multiqc.yaml"
message:
"Compiling a HTML report for quality control checks. Writing to {output}."
shell:
"multiqc {input} -o ../results/ &> {log}"
rule trim_galore:
input:
["../../data/{sample}_1.fastq.gz", "../../data/{sample}_2.fastq.gz"]
output:
- ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"]
+ temp(["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"])
params:
"--paired"
log:
"logs/trim_galore/{sample}.log"
message:
"Trimming using these parameter: {params}. Writing logs to {log}. Using {threads} threads."
conda:
"./envs/trim_galore.yaml"
threads: 8
shell:
"trim_galore {input} -o ../results/trimmed/ {params} --cores {threads} &> {log}"
rule bwa:
input:
fastq = ["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"],
refgenome = config['REFGENOME']
output:
"../results/mapped/{sample}.bam"
log:
"logs/bwa_mem/{sample}.log"
message:
"Mapping sequences against {input.refgenome}"
conda:
"./envs/bwa.yaml"
threads: 8
shell:
"bwa mem -t {threads} {input.refgenome} {input.fastq} > {output} 2> {log}"
# Remove output of last run
rm -r ../results/*
# Run dryrun/run again
snakemake --dryrun --cores 32 --use-conda
snakemake --cores 32 --use-conda
Now when we have a look at the results/trimmed/ directory with:
ls -lh ../results/trimmed/
These fastq files have been removed once Snakemake no longer needs the files for another rule/operation, and we’ve saved some space on our computer (from 12 megabytes to 0.024 megabytes in this directory).
My output:
total 24K
-rw-rw-r-- 1 lkemp lkemp 2.9K Nov 24 16:18 NA24631_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:18 NA24631_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.7K Nov 24 16:18 NA24694_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:18 NA24694_2.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 2.8K Nov 24 16:18 NA24695_1.fastq.gz_trimming_report.txt
-rw-rw-r-- 1 lkemp lkemp 3.0K Nov 24 16:18 NA24695_2.fastq.gz_trimming_report.txt
This become particularly important when our data become big data, since we don’t want to keep any massive intermediate output files that we don’t need. Otherwise this can start to clog up the memory on our computer. And it’s tidy.
4.5 Generating a snakemake report
With Snakemake, we can automatically generate detailed self-contained HTML reports after we run our workflow with the following command:
snakemake --report ../results/report.html
Note. you won’t be able to view this html while it is on the remote server, however after you transfer it to your local computer you should be able to view it
This gives us an interactive version of our directed acyclic graph (DAG).
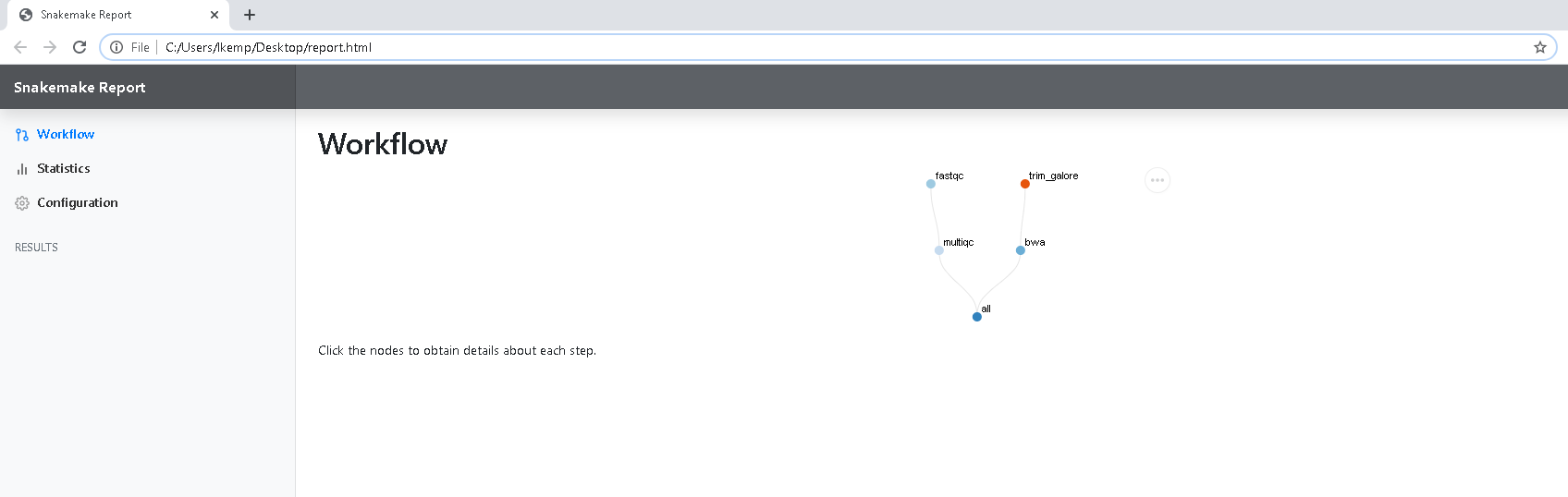
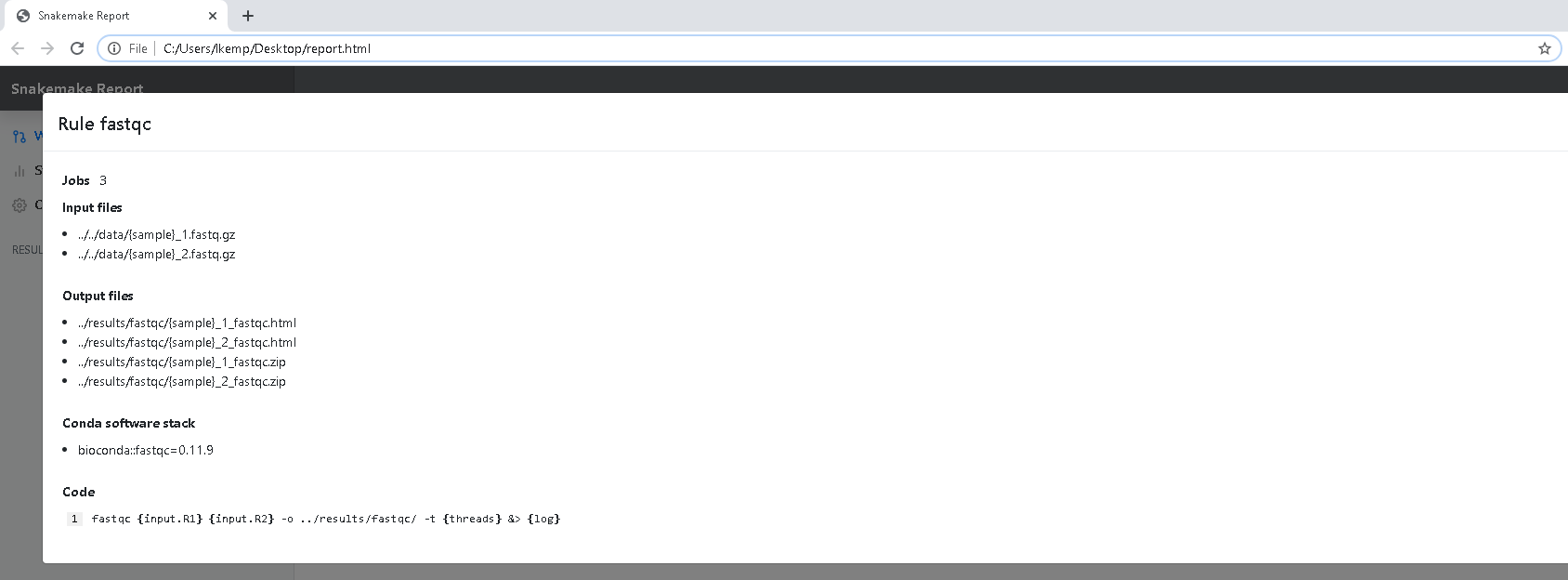
When you click on a node in the DAG, the input and output files are fully outlined, the exact software used and the exact shell command that was run.
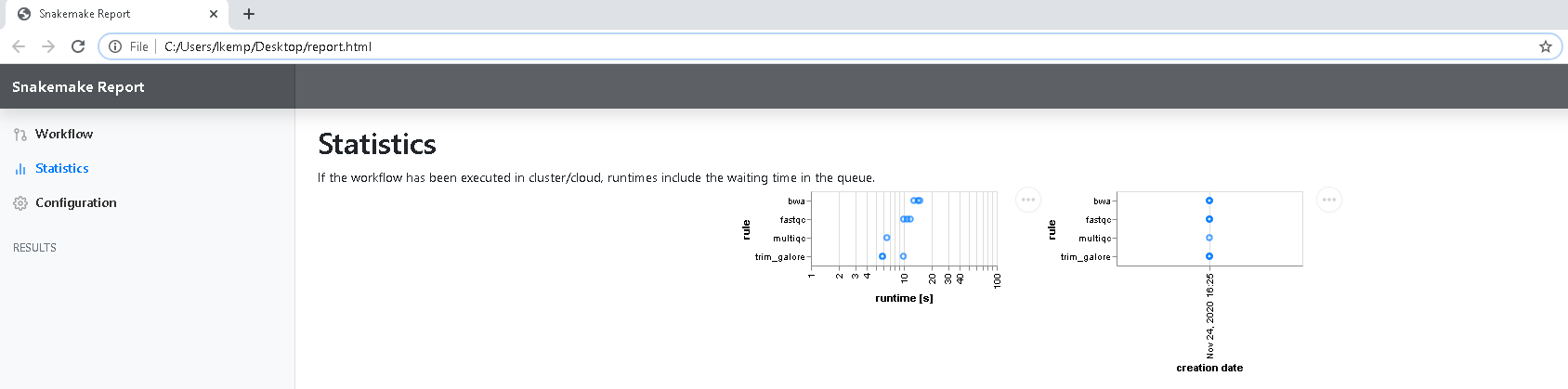
You also are provided with runtime information under the Statistics tab outlining how long each rule/sample ran for, and the date/time each file was created.
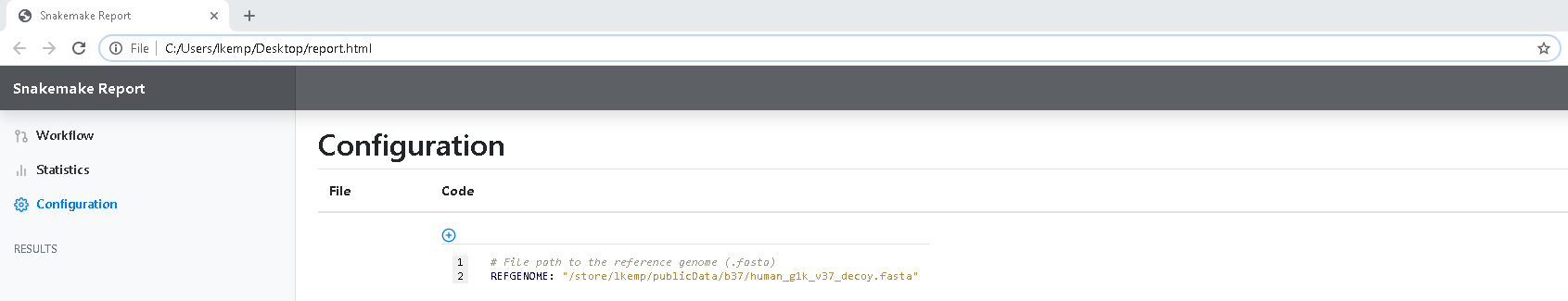
Under the Configuration tab, all of our user configuration is explicitly outlined
These reports are highly configurable, have a look at an example of what can be done with a report here
See more information on creating Snakemake reports in the Snakemake documentation
Takeaways
- Pull out your parameters and put them in
params:directive - Pulling the user configurable options away from the core workflow will support reproducibility by reducing the chance of changes to the core workflow
- Putting your
- Leaving messages for the user of your workflow will help them understand what is happening at each stage and follow the workflows progress
- Mark files you won’t need once the workflow completes to reduce the memory usage - particularly when dealing with big data
- Generate a snakemake report to get a summary of the workflow run - these are highly configurable
Summary commands
Use the parameter directive (params) to keep the parameters and flags of your programs separate from your shell command, for example:
params:
"--paired"
Run your snakemake workflow (using conda to install your software AND with a configuration file) with:
snakemake --cores 32 --use-conda --configfile ../config/config.yaml
Alternatively, define your config file in the Snakefile:
configfile: "../config/config.yaml"
Use the message directive to provide information to the user on what is happening real time, for example:
message:
"Undertaking quality control checks {input}"
Mark temporary files to remove (once they are no longer needed by the workflow) with temp(), for example:
temp(["../results/trimmed/{sample}_1_val_1.fq.gz", "../results/trimmed/{sample}_2_val_2.fq.gz"])
Create a basic interactive Snakemake report after running your workflow with:
snakemake --report ../results/report.html
Our final snakemake workflow!
See leveled_up_demo_workflow for the final Snakemake workflow we’ve created up to this point